Sickle Cell Disease Explained: Causes, Symptoms, And Treatments
All of us know what blood looks like. But that red, slightly viscous, and (sometimes) fear-inducing liquid that oozes out from breaks in our skin (or certain orifices) has more to it than meets the eye.
Blood is part of the cardiovascular system, which is the organ system that functions to transport blood throughout the whole body (per Medical News Today). This system also includes the heart, arteries, veins, and capillaries. According to MedlinePlus, blood is a mixture of red blood cells (RBCs), white blood cells (WBCs), and platelets in plasma. Additionally, the different types of blood cells serve distinct functions from one another. RBCs carry oxygen from the lungs to different areas of the body, WBCs help protect the body from infections and harmful substances, and platelets are responsible for forming clots to prevent excessive bleeding.
Looking at things at a molecular level, every single cell in our body is governed by a genetic code that dictates what type of cell it will be and how it will function. Like the genes of any other cell in the body, blood cells can also become mutated at any point in a person's lifetime. According to the National Human Genome Research Institute (NHGRI), genetic mutations can be acquired (ie, somatic) or inherited (ie, germline). Some mutations are harmless, but others are not. Sickle cell disease (SCD) is one illness that can arise from inherited mutations (via Cleveland Clinic). Here's what you need to know about it.
What is sickle cell disease?
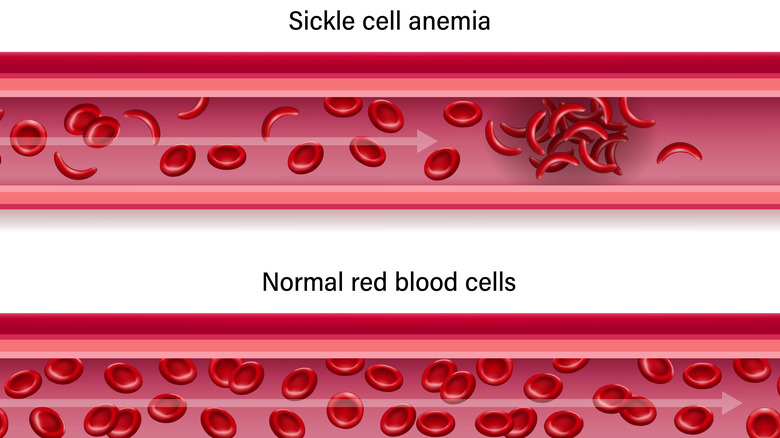
SCD is a condition that affects RBCs. Specifically, it is caused by an inherited mutation that targets hemoglobin, the component of RBCs that contains iron and is responsible for carrying oxygen.
Authors from a 2020 article published in Annual Reviews explain that SCD is a cycle that begins when mutated RBCs are exposed to conditions that require a lot of oxygen. Unlike normal RBCs which are more flexible and are able to adapt, mutated RBCs become rigid and distorted (i.e., sickled), causing them to die prematurely and damage the inner walls of blood vessels. In addition, because of their unnatural shape, sickled cells are also more likely to block vessels and impair blood flow. All of these effects can lead to inflammation (which further narrows the vessels), and is the main explanation behind the many signs and symptoms of SCD.
According to MedicalNewsToday, SCD has many different types. The most common ones include sickle cell anemia (HbSS), hemoglobin SC disease (HbSC), and hemoglobin S-beta-thalassemia disease. HbSS occurs when a child inherits a sickle cell gene from each parent, and is the most severe form of SCD. On the other hand, HbSC occurs when a sickle cell gene is inherited from one parent, and an HbC gene is inherited from the other parent. HbS-beta-thalassemia disease is similar to this in that one parent passes a sickle cell gene to their child while the other parent passes a beta-thalassemia gene.
Sickle cell disease risk factors and statistics

Because SCD is an inherited condition, its biggest risk factor is having parents who have SCD themselves or are carriers of the sickle cell gene. While an SCD carrier doesn't automatically have SCD, they do have sickle cell trait.
Per the National Heart, Lung, and Blood Institute (NHLBI), close to 100,000 people in the United States have SCD, the majority of whom are Black. Specifically, 1 in 13 Black babies will have sickle cell trait, while 1 in 365 Black babies will have SCD. Other races and ethnicities where SCD is also common include Hispanic, Southern European, Middle Eastern, and Asian Indian.
The Centers for Disease Control and Prevention (CDC) adds that SCD commonly runs in families from sub-Saharan Africa, Central America, South America, the Caribbean, Saudi Arabia, India, and the Mediterranean. Per the CDC, death in children from SCD and its related complications significantly dropped since 1999, around the same time the pneumococcal vaccine became available. A 2019 study in the Journal of the American Medical Association reported that in addition to vaccinations, other interventions such as newborn screening, use of antibiotics prior to infections, and use of hydroxyurea have significantly improved the life expectancy of infants and young children with SCD. In fact, about 95% of these children are likely to reach adulthood. Unfortunately, the risk of death and low quality of life remains a problem in adults with SCD, and more research needs to be done.
Sickle cell disease inheritance pattern and screening tests

Inherited disorders can be passed down from parent to child in different ways. In the case of SCD, the Indiana Hemophilia & Thrombosis Center (IHTC) illustrates that if both parents each have one normal hemoglobin gene and one sickle cell gene, their child has a 25% chance of inheriting normal genes (no disease), a 50% chance of inheriting one normal gene and one sickle cell gene (sickle cell trait), and a 25% chance of inheriting two sickle cell genes (SCD). These odds increase if one or both parents have SCD instead of sickle cell trait, which is why it is very important to screen for the presence of the sickle cell gene in newborns, as well as in couples who are considering pregnancy.
Per Healthline, testing for SCD or sickle cell trait only requires a blood draw. The blood sample is analyzed for the presence of sickled cells, which look like crescent moons. In contrast, normal red blood cells are round with a central depression, akin to donuts. Aside from newborns, SCD should be screened in immigrants and children who have never been tested, and anyone experiencing symptoms. The NHLBI adds that aside from a blood test, a genetic test that looks for whether or not you have the gene can also be done. If a couple is pregnant and they want to know if their unborn baby has SCD, a placental tissue or amniotic fluid sample can be taken from the mother as early as 8 to 10 weeks.
Common symptoms of sickle cell disease

The signs and symptoms of SCD are largely due to the distortion of RBCs and their tendency to block blood vessels and cause inflammation. When RBCs take on a sickle shape, oxygen is unable to bind efficiently to hemoglobin, blood flow to other parts of the body becomes impaired, and the RBCs become more fragile (via WebMD). This is followed by a significant number of RBCs dying (hemolysis), which in turn leads to anemia and jaundice.
Anemia occurs when there aren't enough RBCs circulating in the blood. It can cause a variety of symptoms, including difficulty breathing, lightheadedness, fast heartbeat, tiredness, pale skin, delayed growth, and delayed puberty. On the other hand, jaundice occurs when too much hemoglobin is being broken down, which is a consequence of rapid RBC turnover. It can be seen as yellowing of the skin or the white parts of the eyes.
Another common symptom of SCD is swelling of the hands and feet (dactylitis or sausage fingers) caused by impaired blood flow (per MedicalNewsToday). This happens more commonly in infants and young children between 6 months old to 4 years old. It may be accompanied by fever, pain, and increase in white blood cell count. Dactylitis can also be seen in other conditions such as arthritis, tuberculosis, and syphilis.
Sickle cell disease diagnosis
According to the American Red Cross, screening newborns for the presence of the sickle cell gene did not become mandatory in the United States until 2006. Unfortunately, although there is a clear benefit to screening, the Health Resources & Services Administration (HRSA) says that most U.S. states allow parents to refuse newborn screening for their babies. The law varies from state to state, though: Some strictly implement it, others allow refusal due to religious beliefs, but there are also states that allow refusal for any reason. Therefore, for anyone who is not known to have SCD, a doctor can provide a diagnosis by obtaining a detailed medical history and ordering certain tests.
A medical history typically includes a person's family history, as well as their previous and/or current symptoms (if any). Healthline states that if a doctor suspects that you have SCD, they most likely will order a blood draw (the same one used for screening), and the blood sample may be subjected to different types of tests. These may include measuring the amount of RBCs in the blood sample, looking for the presence of sickled cells, and testing the solubility of hemoglobin. In addition to this, hemoglobin electrophoresis is a test that measures hemoglobin levels in a sample. It can also detect and measure the amount of abnormal types of hemoglobin (e.g., HbS, HbC). It is done to confirm the diagnosis.
Treatment of sickle cell disease
SCD is a chronic condition that requires regular monitoring of symptoms. Although a bone marrow (stem cell) transplant is known to cure some people, it is associated with risks (including death), and the choice to undergo this procedure depends on how severe the disease is and whether or not there is an available, compatible donor (per Johns Hopkins Medicine). It is performed only by specially trained doctors in specialized clinics.
Medications are available to prevent and manage symptoms. Hydroxyurea is one of the most common medicines prescribed for SCD (via Mayo Clinic). It helps reduce the number of pain episodes, and may even decrease the need for blood transfusions and admissions to the hospital. Unfortunately, hydroxyurea can lower a person's white blood cell and platelet counts, increasing their risk of infections and excessive bleeding. It is also harmful to babies in the womb, so women who can get pregnant and men who are sexually active need to have birth control while taking hydroxyurea.
Other medicines that can be used to manage SCD include L-glutamine powder, crizanlizumab, voxelotor, and narcotics (per Mayo Clinic). These are oral or injection drugs that help relieve pain, reduce the number of pain episodes, lower the risk of anemia, and improve blood flow. They each have their own side effects, and some of them are not meant to be taken by children.
Vaso-occlusive crisis in sickle cell disease
Vaso-occlusive crisis (VOC) is the most common finding seen in people with SCD (via StatPearls). Responsible for around 95% of hospitalizations related to SCD, it is one of the most common causes of death in SCD (alongside acute chest syndrome). VOC causes sudden and intense episodes of sharp, throbbing pain, often felt in the lower back, joints, and limbs. The pain is severe enough to warrant admission to the hospital, usually lasting a week per episode.
Authors from a 2020 article in the European Journal of Hematology explain that the pain felt during a VOC is unlike any other. It is caused by a combination of factors, including occlusion of blood vessels by sickled cells, reduced or possibly absent blood flow, inflammation, and tissue damage. Unfortunately, people with SCD are naturally in constant pain due to the many pathologies that can occur throughout their body. They may have pain caused by lack of circulation in their bones, leg ulcers, chronic bone infections, and side effects from their medications. Sometimes, they may even experience pain without any identifiable, underlying cause.
To provide the proper treatment, prevent further complications, and avoid the possibility of overlooking a potentially life-threatening pathology, it is important to distinguish VOC from the other causes of pain (per StatPearls). Unfortunately, this can only be done subjectively, since there are no tests or procedures available that can measure the quality and severity of pain a person is experiencing.
Splenic sequestration crisis in sickle cell disease

The spleen is located in the upper left area of the abdomen, and is responsible for filtering blood by removing dead blood cells and bacteria from the body. The Texas Department of State Health Services (DSHS) states that splenic sequestration crisis (SSC) happens when blood vessels exiting the spleen get blocked by sickled cells, causing blood to accumulate. When this happens, the spleen enlarges (so much so that it can easily be felt by palpating the area where it is located), hemoglobin and hematocrit levels fall, and bacteria does not get removed from the blood. SSC can sometimes be painful, and can often present with weakness, irritability, unusual sleepiness, pale skin, and fast heartbeat. SSC is commonly seen in infants and children between 2 months old and 4 years old, and is the period where people with SCD are at greatest risk of infections.
Minor splenic sequestration episodes are sometimes triggered by viral infections and are characterized by a slightly enlarged spleen, and small reductions in hemoglobin and platelet levels (via the IHTC). Although the spleen becomes nonfunctional in people with SCD by 5 years old (thus making them no longer at risk of SSC), if a person has more than one episode, removing the spleen might be the best way to avoid future occurrences. On the contrary, major episodes are fatal and can result in death within hours. Prompt treatment with fluids and blood transfusions is necessary. Removal of the spleen (if needed) can be done once they are stable.
Other complications that can occur in sickle cell disease
SCD can affect virtually any organ in the body because of its tendency to cause clogging in blood vessels. Acute chest syndrome (ACS) is a devastating, life-threatening complication of SCD. Together with vaso-occlusive crisis (VOC), ACS is one of the primary causes of death, and the second most common reason for hospitalizations, in people with SCD. Similar to what happens in VOC and splenic sequestration crisis (SSC), ACS occurs when blood vessels in the lungs get blocked by sickled cells, causing a person to have sudden chest pain, difficulty breathing, coughing, and fever (via Cleveland Clinic).
Stroke is also a common complication of SCD (11% in people younger than 24 years old, 24% in people younger than 45 years old). It can lead to brain damage and other permanent consequences (e.g., changes in speech and mobility), and having one episode increases a person's chance of having another (per Johns Hopkins Medicine). A person with SCD may also develop frequent bacterial infections, priapism (painful erections that can last for hours or longer), pulmonary hypertension (presenting as fast pulse, shortness of breath, and fainting), and leg ulcers. Chronic kidney disease is also common (around 30%), and it causes progressive symptoms such as fatigue, swelling, urinary problems (e.g., needing to pee frequently, blood or foam in pee), and high blood pressure. SCD can also affect the eyes (e.g., detachment of the retina) and cause several vision problems (via Cleveland Clinic).
How to prevent sickle cell disease complications
Complications are common (and sometimes deadly) in SCD, and treating these complications when they occur is often not enough. In people with SCD, preventing complications is key to preserving their health and quality of life.
According to the CDC, the best way to reduce the risk of infections in SCD is through vaccinations. Aside from routine childhood vaccines and yearly flu shots, children and adults with SCD should also receive the pneumococcal vaccine and any other vaccines recommended by their doctor. Doctors can also prescribe antibiotics to prevent infections. Specifically, children with SCD should be given penicillin daily until they reach 5 years old. The earlier they begin penicillin treatment, the more effective it is at protecting against infections. To prevent vision problems, regular visits to an eye doctor is necessary. They will be able to identify if there are any abnormalities in your eyes (specifically the retina), and treat them before they get worse.
Blood transfusions are often done in people with SCD to prevent (and treat) strokes and sickle cell anemia (per Mayo Clinic). The procedure is the exact opposite of giving blood. A needle is inserted in a vein, which is then replaced by a small tube. The tube is attached to a bag of compatible donor blood, and this blood will slowly flow through the tube and into the vein. Blood transfusions increase the number of RBCs, and temporarily replaces sickled cells with normally shaped RBCs.
What you can do at home

What you do at home on a daily basis is also crucial to controlling and preventing SCD symptoms. According to Healthline, you should practice healthy eating by incorporating the right amount of fruits, vegetables, and grains in your diet to help your body produce more red blood cells. You should also drink plenty of fluids to prevent dehydration (a common trigger for sickle cell crisis), exercise regularly, and take folic acid supplements if prescribed by your doctor. Although folic acid is often given to people with SCD with the intention of preventing folate deficiency, authors of a 2018 review in the Cochrane Library state that the benefit of doing this has yet to be clearly proven.
The CDC also advises people with SCD to consciously avoid things that can be potential triggers of symptoms. These include extreme temperatures, activities that involve being in high-altitude places (e.g., flying, hiking), places where the air is thin (e.g., mountains, ski resorts), and situations that can deplete your oxygen level (e.g., extreme exercise, athletic training). Protect yourself from infections by avoiding close or direct contact with people who are sick or have been exposed to a contagious illness. Observe good hygiene by washing your hands frequently with soap and water, and practice proper food handling. Avoid smoking (as this can trigger pain crisis episodes), and be extra cautious when taking medication not prescribed by your doctor, as some of them may be harmful to your kidneys (via Mayo Clinic).
Sickle cell disease versus sickle cell trait

A person has sickle cell trait (SCT) if they inherit only one copy of the sickle cell gene from one parent. They are not considered to have SCD. Healthline explains that if one or both parents have SCT, each one of their children has a 50% chance of having SCT.
According to the CDC, people with SCT rarely experience symptoms. If they do, the symptoms and complications are similar to that of sickle cell disease. It is important to note that people with SCT are more prone to heat stroke and muscle breakdown compared to those without the disease. Similar to SCD, people with SCT should avoid getting dehydrated and should be very careful if they are doing anything that is physically draining (e.g., intense exercise). A diagnosis of SCT can be confirmed using a blood test.
Interestingly, the American Red Cross states that SCT is believed to have originated in Africa thousands of years ago as a way for the body to fight against malaria, a contagious parasitic disease spread via a mosquito bite. This theory is backed up by the fact that SCT is most prevalent among people with African backgrounds, and that people with SCT are actually somewhat protected against malaria. A study by the CDC showed that children who had SCT had greater survival rates against malaria compared to those without SCT and those who have SCD. Children who had SCD surprisingly had worse survival rates compared to those without it.
Similarities and differences between sickle cell disease and thalassemia
Sickle cell disease and thalassemia are both inherited RBC conditions caused by a mutation in hemoglobin (via WebMD). Their presentations and management are similar to one another. Hemoglobin is made up of alpha and beta proteins. Four genes are assigned to alpha proteins (two from each parent), and two genes are assigned to beta proteins (one from each parent). If only one alpha gene is abnormal, you are considered to be a carrier of alpha-thalassemia. If two or more alpha genes are abnormal, you have alpha-thalassemia and the severity of your symptoms will largely depend on how many genes are affected. On the other hand, one abnormal beta gene will cause mild disease, while two abnormal beta genes will cause severe beta-thalassemia. In general, the greater the number of abnormal genes there are, the worse the condition is.
According to a 2022 article (via StatPearls), signs and symptoms of thalassemia may include pale or yellow skin, anemia, changes in appearance ("chipmunk face"), abdominal pain caused by gallstones, enlarged liver and spleen, and slow growth. Similar to SCD, management of thalassemia includes frequent blood transfusions. However, this can lead to too much iron in the blood, which can also cause a variety of symptoms such as bronzing of the skin, irregular heart rhythm, liver failure, and hormone imbalances. To avoid this, part of the treatment for both thalassemia and SCD involves removing the excess iron in the blood. This can be done using iron chelators like deferoxamine.
Sickle cell disease and pregnancy

Authors of a 2019 article in the Mediterranean Journal of Hematology and Infectious Diseases state that before newborn screening began and preventive strategies against SCD were made, there was a high risk of death (of both mother and baby) during pregnancy. Although this has decreased significantly, women with SCD who get pregnant are still at a higher risk of complications compared to women who do not have SCD.
Pregnancy and SCD exert harmful effects on one another. Normal changes in pregnancy such as increased viscosity of blood and hypercoagulability (increased tendency of blood to clot) can increase the severity of acute chest syndrome episodes and vasoocclusive crises. Meanwhile, abnormal RBC structure and impaired blood flow in SCD can increase the risk of eclampsia and preeclampsia.
Despite these risks, many women with SCD are still able to have healthy pregnancies (per Norton Healthcare). The most important thing is to have frequent check-ups with a qualified doctor to ensure that the mother's SCD is controlled and the baby inside the womb is growing normally. If you are taking hydroxyurea, your doctor will discontinue this drug temporarily while you are pregnant and breastfeeding. A vaginal delivery can be done, but because a mother can lose a lot of blood during and shortly after birth, it is very important to keep them hydrated to prevent a pain crisis from happening.
Life with sickle cell disease

People with SCD are living past their 50s, which is significantly longer than before. If you or someone you know has SCD, it is important to recognize what symptoms warrant a trip to the emergency room. These include extreme weakness; shortness of breath; dizziness; abnormal heartbeat; high-grade fever; chest pain; numbness; confusion; changes in speech, vision, and mobility; and prolonged, painful erections (per Cleveland Clinic).
The CDC advises people with SCD to find healthcare providers who are knowledgeable and experienced in managing the disease, and have check-ups with them regularly. Starting from birth to 12 months old, babies with SCD should be seen by their doctor every 2 to 3 months. Children between 1 to 2 years old should be seen at least every 3 months, and anyone over 2 years old should see their doctor at least once a year. It can also be helpful to look into clinical trials to check if there are any new treatment options worth exploring with your doctor, and join support groups or organizations to learn more about SCD and get assistance if you need it.
The American Sickle Cell Association (ASCAA) provides all kinds of services to people and families with sickle cell trait or SCD, collecting and consolidating statistical and case data on SCD for the purposes of helping research studies. Meanwhile, the Sickle Cell Disease Association of America (SCDAA) supports several organizations, research studies, and health education involved in treating SCD and raising SCD awareness.