Everything You Need To Know About Cystic Fibrosis

It goes without saying that we, quite literally, owe our lives to our parents. We wouldn't exist if it weren't for them. Biologically speaking, our genes are an equal mixture of genetic material from each parent (via MedlinePlus). Unfortunately, as much as we would have wanted to receive only the best genes our parents had to offer, nature just doesn't work that way. Some of us inherit genetic mutations that cause hereditary diseases, the most common of which include hemophilia, sickle cell disease, Huntington's disease, and cystic fibrosis (via Healthgrades).
Among the many genetic health conditions that can be passed down from parent to child, cystic fibrosis is one of the diseases that is screened in newborns. According to the Health Resources & Services Administration (HRSA), newborn screening is required by law to be offered throughout the United States, but some states allow parents to refuse screening. It's important to understand that screening is an important tool that parents should utilize for their baby's health. It provides an opportunity for potentially disabling or life-threatening conditions to be identified and treated early, which can decrease or even eliminate the health consequences that are associated with them. All it takes is a few drops of blood, a hearing test, and a sensor that screens the baby for certain heart conditions. The Cystic Fibrosis Foundation (CFF) states that in people with cystic fibrosis, early diagnosis and treatment can lead to improved growth, better lung function, and longer survival.
What is cystic fibrosis?

Cystic fibrosis (CF) is a long-term, progressive medical condition that affects many systems in the body. According to the CFF, it occurs in people who have a mutation in the gene that codes for a protein called the cystic fibrosis transmembrane conductance regulator (CFTR). Mutations in the CFTR gene can lead to CFTR proteins that are dysfunctional, deficient in numbers, or completely absent. The CFTR protein is responsible for maintaining salt (i.e., sodium chloride) and water balance in certain areas of the body. When this balance is disrupted, the chloride in salt gets trapped inside cells, and water is unable to move through the surfaces to hydrate them. This causes a buildup of thick and sticky mucus, which in turn leads to the signs and symptoms of CF.
Smith Allergy & Asthma explains that mucus is an important substance that lines the surfaces of many organs in the body, such as the nose, throat, lungs, stomach, and intestines. It provides lubrication and creates a protective barrier against harmful substances like microorganisms and environmental irritants. If you ever wondered why you get a runny nose when you have allergies or a cold, that's your body's way of responding to foreign invaders — by increasing mucus production. Mucus should be adequately produced and have the right consistency in order for it to effectively serve its purpose. The abnormal consistency of mucus produced by people with CF makes them vulnerable to infections, plugging of airways, and organ damage (via MedlinePlus).
Who is at risk of developing cystic fibrosis?

Cystic fibrosis is inherited by a child from their parents (via CFF). Both parents must have a copy of the mutated CFTR gene. A child who inherits one copy is considered a CF carrier, while a child who inherits a copy from each parent will have CF. If two CF carriers conceive, their child has a 25% chance of having CF, a 50% chance of being a carrier, and a 25% chance of being neither. If a person with CF and a CF carrier have a child, there is a 50% chance that the child will have CF, and a 50% chance that the child will be a carrier. If a person with CF has a child with a person who is neither a carrier nor has the disease, the child will always be a carrier (via Texas Children's Hospital). It's important to know that having a child with CF (or one without) does not change your risk of having another child with CF.
Family history is a significant risk factor, but some ethnicities have a higher risk compared to others. According to Healthline, the risk of carrying the mutated CFTR gene is highest for Caucasians (1 in 29), followed by Hispanics (1 in 46), African-Americans (1 in 65), and Asians (1 in 90). The chance of having a child with CF is also highest for Caucasians (1 in 2,500-3,500), followed by Hispanics (1 in 4,000-10,000), African-Americans (1 in 15,000-20,000), and Asians (1 in 100,000).
Symptoms of cystic fibrosis

Cystic fibrosis can present with many different symptoms. They are not always the same for everyone, and the severity of symptoms varies as well. Stanford Medicine states that early symptoms of cystic fibrosis seen in babies or young children include blocked intestines at birth, unusual bowel movements (e.g., persistent diarrhea, bulky and smelly stools, constipation), salty skin or sweat, irregular eating patterns, weight loss, lack of energy, breathing problems, unusual tiredness, persistent cough, and wheezing. As the disease progresses, these symptoms can worsen and lead to coughing up mucus and blood, inability to exercise, prolapsed rectum, nasal or sinus polyps, clubbing (i.e., rounding or flattening of the fingers and toes due to lack of oxygen in the blood), and infertility.
Johns Hopkins Medicine adds that people with CF can also experience malnutrition, poor growth, frequent respiratory infections, and permanent lung disease. CF can cause the lungs to collapse, the right side of the heart to enlarge, and the pancreas to become inflamed (i.e., pancreatitis). Other medical conditions also associated with CF include sinusitis, liver disease, diabetes, gallstones, and specifically in males, congenital bilateral absence of the vas deferens (CBAVD), which can lead to the sperm canal becoming obstructed. Because symptoms of CF are nonspecific and can also be seen in other health conditions, you should speak with your doctor if you suspect that you or your child may have CF.
Cystic fibrosis and the respiratory system
The respiratory system is made up of many organs and specialized tissues including the nose, mouth, sinuses, pharynx (throat), larynx (voice box), trachea (windpipe), diaphragm, and lungs. They work together to help you breathe, talk, smell, warm and humidify the air you breathe in, as well as protect you from infections and irritants (per Cleveland Clinic).
The respiratory system is the most commonly affected organ system in cystic fibrosis (via Cystic-Fibrosis.com). Mucus normally lines the surfaces of respiratory organs, which provides lubrication and functions to trap harmful substances so that they can be expelled from the body through coughing and sneezing. Interestingly, researchers of a 2012 study published in the Current Biology journal found that mucin, the major component of mucus, causes bacteria to move faster, which in turn prevents them from attaching to underlying surfaces and causing disease. However, people with cystic fibrosis have mucus that is thick and sticky in consistency, which can clog the airways and is a welcoming environment for viruses, bacteria, and fungi to grow and multiply (per Cystic-Fibrosis.com). Because of this, people with cystic fibrosis frequently have infections and inflammation of the airways, and over time can lead to permanent lung damage and other complications. In addition, taking too many antibiotics can cause antibiotic resistance, which leads to future infections that can no longer be treated with medicines.
Cystic fibrosis and the gastrointestinal system
The Cleveland Clinic explains that the gastrointestinal (GI) system is a continuous network of hollow organs that are connected to one another. It begins at the mouth, ends at the anus, and in between are the esophagus, stomach, liver, gallbladder, pancreas, and intestines. It is responsible for processing the food and beverages we consume so that the nutrients they contain can be readily absorbed by our body and used for energy, growth, and repair.
According to the Children's Hospital of Philadelphia (CHOP), the pancreas is the most commonly affected GI organ in cystic fibrosis. Similar to mucus, secretions from the pancreas become thick, which can block the pancreatic ducts. The pancreas is responsible for secreting substances that aid in digestion, so if the ducts get clogged, these substances are unable to exit the pancreas. This results in problems with absorbing fat, protein, and fat-soluble vitamins, which in turn leads to greasy, bulky, and foul-smelling stools. People with cystic fibrosis may also develop type 1 diabetes. This happens when insulin becomes deficient because of damage to the pancreas. In fact, the incidence of diabetes in people with CF is around 35% in their 20s, and more than 40% in their 30s. Other parts of the GI system can also be affected in cystic fibrosis and cause stomach pain, liver disease, gallstones, and bowel obstruction.
Cystic fibrosis and the reproductive system

The reproductive system is responsible for all things related to sexual reproduction. The anatomies of the male and female body are designed in a way that complements each other in order to create life (via Winchester Hospital). Many organs comprise the reproductive system, but in cystic fibrosis, the most commonly affected organs are the vas deferens in males and the cervix in females.
According to a 2019 article published in the Journal of Cystic Fibrosis, more than 98% of men with CF are infertile due to being born without a vas deferens. The vas deferens is a pair of tubes that serves as a passageway for sperm to travel from the epididymis (coiled tube connected to the testis) to the ejaculatory duct (via Cystic Fibrosis News Today). Some men may also have small or absent seminal vesicles (organs that secrete the fluid in semen) and missing portions of the epididymis. All of these contribute to infertility because although sperm production and libido are normal, the sperm has no way of exiting the testes because the tubes are nonexistent.
In contrast, women with CF can have thickened cervical mucus that prevent sperm from effectively reaching the egg for fertilization. Women may also experience irregular or delayed menstrual periods, or not have them at all. In some cases, complications from other symptoms of the disease (e.g., malnutrition) can also impair ovulation. All in all, these factors increase the risk of infertility in women with cystic fibrosis.
How is cystic fibrosis diagnosed?

Cystic fibrosis is often diagnosed during infancy or childhood. Newborns are screened for the disease (as well as other health conditions) by placing a few drops of their blood on a special card (per the HRSA). The card is analyzed for cystic fibrosis by measuring the amount of a substance called immunoreactive trypsinogen (IRT). IRT is increased in babies with cystic fibrosis (and also sometimes in CF carriers, or in babies with other health issues), and an abnormal result in newborn screening requires further testing for the disease.
A genetic test using a blood sample can also be done to look for mutations in the CFTR gene. It can identify specific mutations majority of the time. There are over 2,000 identified mutations in this gene alone, but the most common one is the deltaF508 mutation (via the American Lung Association). Genetic tests can be performed in babies who need additional testing, as well as in couples who are planning to get pregnant and want to know if they're carriers of the disease.
The Cleveland Clinic adds that a nasal potential difference (NPD) test can be used to measure the transfer of ions across the lining of your nose. An abnormal result may help in confirming the diagnosis. Other supportive tests include chest and sinus X-rays, lung function tests, and sputum culture, but the most reliable test in diagnosing cystic fibrosis by far is the chloride sweat test.
The chloride sweat test
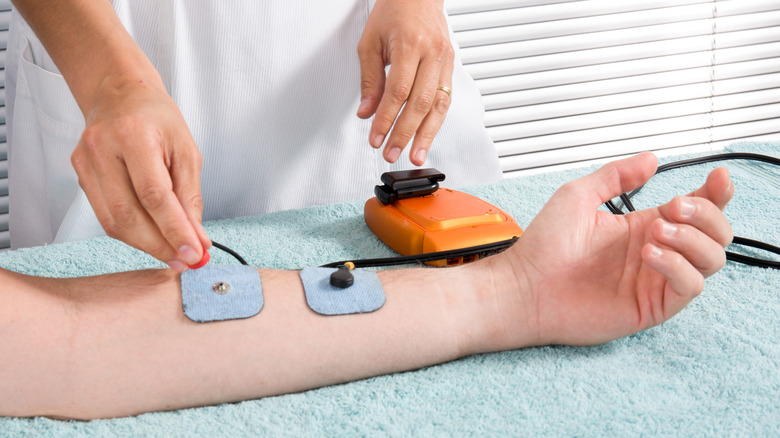
The sweat test measures the amount of chloride in sweat, which is expected to be higher than normal in people with cystic fibrosis (per the CFF). It can be performed for anyone over two days old, but it is recommended that babies who were identified as potentially having CF through newborn screening or prenatal genetic testing should have it done by four weeks old.
Nemours KidsHealth explains the sweat test process. A designated area on the arm is cleansed and dried. Two electrodes are strapped around the limb, with one electrode containing pilocarpine (a drug that causes sweat glands to produce sweat), followed by a little electrical stimulation to help the skin absorb the medicine. A warm or tingling feeling may be felt during this time. The electrodes are then removed, and the skin is cleansed again. At this point, the area of the skin that was stimulated should be producing sweat. The sweat is collected using a special device for 30 minutes, and is then sent to a laboratory for analysis. It's normal for the skin to stay red or continue sweating for several hours after.
A result of less than 30 mmol/L means that CF is unlikely (via WebMD). CF is likely if the result is greater than or equal to 60 mmol/L. If the results are inconclusive (30-59 mmol/L), or not enough sweat was produced (which often occurs in newborns), a repeat sweat test should be performed.
Medications for cystic fibrosis

Cystic fibrosis is a lifelong condition that cannot be cured, but its symptoms and complications can be managed (per the CFF). Antibiotics can be given to both treat and prevent lung infections. People with cystic fibrosis take oral or inhaled antibiotics as part of their daily treatment. For acute infections, antibiotics may need to be administered intravenously. Anti-inflammatory drugs like ibuprofen and corticosteroids reduce inflammation in the airways and can be given to relieve symptoms. Similarly, mucus thinners such as hypertonic saline can provide relief by making it easier to cough out thick mucus in the lungs. Bronchodilators (e.g., albuterol) are inhaled drugs that relax airway muscles to allow more air to pass through. These can relieve symptoms and help other drugs work better. CFTR modulators correct the defective protein produced by the mutated CFTR gene. These drugs are effective only in people with specific mutations. CFTR modulators are effective as long as they're in your system, so you have to continuously take them every 12 hours or as prescribed by your doctor.
In 2019, the Food and Drug Administration (FDA) approved Trikafta, a combination therapy that targets the abnormal CFTR protein. It can be used in people 12 years and older who have at least one deltaF508 mutation, which is approximately 90% of people with CF. As of September 2022, the FDA also approved Orkambi, a CFTR modulator that can be used for children 1-2 years old who have two deltaF508 mutations (via CFF).
Other treatment options for cystic fibrosis

According to the Mayo Clinic, aside from medications, people with cystic fibrosis may need pancreatic enzyme supplements to help with absorption of nutrients, and stool softeners to prevent constipation and bowel obstruction. They may also need to undergo certain procedures caused by complications from the disease. These can include nasal and sinus surgery, oxygen therapy, bowel surgery, lung transplant, liver transplant, or having a feeding tube.
Airway clearance techniques (ACTs) are taught to people with CF and parents who have infants or toddlers with CF. Cystic-Fibrosis.com lists the numerous techniques that can be done, which include coughing or huffing, postural drainage (letting gravity drain mucus by assuming different positions), chest percussion (clapping the chest with a cupped hand), positive expiratory pressure therapy (involves breathing through a mask), active breathing techniques, autogenic drainage (using different speeds and depths of breathing to move mucus), and vest therapy (a vest is wrapped around the body and vibrates at a high frequency to help loosen mucus).
Other things that can aid in managing cystic fibrosis include proper nutrition, adequate exercise, updated vaccinations, and stress management. According to Healthline, since people with cystic fibrosis have problems with nutrient absorption, it's important that they follow a diet that accounts for these deficiencies. This entails increasing the amount of calories, protein, fat, salt, fiber, calcium, iron, zinc, and antioxidants in their diet.
Complications from cystic fibrosis

Cystic fibrosis can be life-threatening if it isn't being managed well (per Mayo Clinic). Complications may arise in any of the multiple organ systems it affects. It can affect the bones and cause osteoporosis, cause electrolyte imbalances and dehydration, and increase the risk of having mental problems like anxiety and depression. In the respiratory system, it can cause permanent lung damage, prolonged infections that are resistant to treatment, coughing up blood, fleshy growths inside the nose, and lung collapse. Over time, the lungs may become so damaged to a point where they can no longer function properly. When this happens, respiratory failure develops — this is the most common cause of death in those with cystic fibrosis. Bowel obstruction can occur in the GI tract and is a medical emergency that requires urgent treatment. Liver disease like fatty liver and cirrhosis may also develop, and people with CF may eventually need a liver transplant.
According to the CFF, people with cystic fibrosis are 5 to 10 times more likely to develop colorectal cancer compared to the general population. In addition, people who have had a solid organ transplant are 20 times more likely to develop colorectal cancer. Normally, screening for colorectal cancer begins at 50 years old. But because of the risks, people with CF should start screening at 40 years old, and those with a history of a solid organ transplant should begin at 30 years old.
Genetic screening for cystic fibrosis

Cystic fibrosis is not a preventable disease. However, couples who are planning to have a child may undergo genetic screening to determine if they are carriers, so that they know what their chances are of having a child with CF. According to the American College of Obstetricians and Gynecologists (ACOG), screening is completely voluntary and can be done before or after pregnancy. It can be done in three ways: ethnic-based screening (tests that check for diseases that you're at risk for based on your ethnicity), expanded carrier screening (tests for many disorders at once), or limited testing for specific disorders. Choosing to get screened before pregnancy provides more options if the results show that you and your partner are carriers. These include accepting the risks and getting pregnant, deciding not to have children, opting for adoption, trying in vitro fertilization (IVF), or making use of a sperm or egg donor. If you're already pregnant, prenatal testing can determine if your child has cystic fibrosis or if they are a carrier.
If you are a carrier of, or have, cystic fibrosis, it's important that you tell your partner so that you can make a joint decision on how to proceed. You should also consider telling your family, since they may be carriers or have the disease as well. But know that it is still your choice whether or not to disclose this information, and you're not required to share this with anyone if you do not want to.
Living with cystic fibrosis

Cystic fibrosis is often diagnosed in children less than 2 years old, and more people with CF are now being identified early in life thanks to newborn screening. Cystic Fibrosis News Today chronicles how life is like when you have CF. As a child grows older, existing symptoms may worsen, and new ones may develop. Despite the risks, a diagnosis of cystic fibrosis should not prevent a child from attending school. But there are some challenges that can come with this, both physical and mental. Physically, a child with CF is more prone to infections and other related conditions, which can cause them to miss school due to frequent hospitalizations. Mentally, a child with CF may struggle with body image issues or bullying. For these reasons, some parents opt for home-schooling instead. As the child grows up, they learn to take care of themselves and become responsible for watching over their own health. This means listening to their bodies, keeping up with doctor's appointments, and making sure to regularly take their prescribed medications.
In the past, people with cystic fibrosis rarely survived into adulthood. But because of advancements in the medical field, people with CF now live up to 35 to 40 years old on average, and some even older than that (via Healthline). According to Cystic Fibrosis, people with CF can live up until their 40s before it becomes likely they will need a lung transplant, which can add around 8.5 more years to life.
The Cystic Fibrosis Foundation

The Cystic Fibrosis Foundation (CFF) was established in 1955 by a group of parents who wanted to raise awareness on cystic fibrosis and work together to fight the disease. Their children had CF, and during that time very little was known about it. Six years later, the CFF successfully opened two accredited treatment centers that were dedicated for people with cystic fibrosis. By 1966, the CF registry containing health information on patients with cystic fibrosis was created, and up to this day remains to be a top resource that is being used for CF research.
The CFF funded a team of researchers who in 1989 discovered the mutated CFTR gene and protein. Four years later, the FDA approved the first drug that was made specifically for cystic fibrosis. Fast forward to today, the foundation continues to fund research dedicated to understanding CF and finding a cure, open specialized clinical research centers, raise awareness through advocacy programs and meetings with elected public officials, and provide support to the CF community.
The CFF now has over 130 accredited CF centers, and more than 100 programs dedicated to treating adults with cystic fibrosis. Life expectancy of people born with CF has doubled over the past 30 years, and it is the CFF's mission to find a cure for cystic fibrosis and give people with CF the chance to live a long and fulfilling life.